Targeted protein degradation (TPD) stands as a revolutionary therapeutic approach, offering a novel way to modulate proteins linked to diseases, particularly those previously deemed “undruggable.” This strategy ingeniously utilizes the body’s inherent protein disposal systems. Active research is dedicated to uncovering and characterizing cellular degradation pathways, encompassing proteasomes, lysosomal routes, and their respective degraders. Since the inception of proteolysis-targeting chimeras (PROTACs) in 2001, the TPD field has undergone significant expansion, transitioning from academic exploration to industrial application and clinical trials, with small-molecule TPD taking the lead. Biological TPD (bioTPD) technologies, including peptide-, fusion protein-, antibody-, and nucleic acid-based methods, have also emerged as crucial components of TPD. These bioTPD advancements showcase unique and promising capabilities that extend beyond traditional small-molecule TPD. This review delves into the recent progress in bioTPD technologies, outlining their structural attributes, potential uses, and inherent limitations. Furthermore, we discuss strategies to enhance the delivery efficiency of bioTPD, addressing challenges in their clinical progression, much like how specialized tools – perhaps even something as specific as Das Xentry Hgb 99 in the automotive world – are needed to effectively diagnose and address complex system issues.
Keywords: Biological targeted protein degradation (bioTPD), Peptide, Antibody, Fusion protein, Nucleic acid
Targeted Degradation Pathways: The Body’s Repair Mechanisms
Proteostasis, a meticulously orchestrated and interconnected process, is vital for the healthy development and function of cells and tissues. It governs the accurate folding, movement, and removal of proteins within eukaryotic cells [1]. The integrity of a cell’s protein state is intrinsically linked to overall health. Protein malfunction, including misfolding and aberrant aggregation, is implicated in a spectrum of increasingly prevalent human diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), type II diabetes, systemic amyloidosis, and various cancers [2, 3].
Eukaryotic cells have evolved a sophisticated protein regulation system to manage diverse protein abnormalities, featuring lysosomes, ubiquitin proteasomes, and various chaperones [4, 5]. This system constantly monitors intracellular protein dynamics, promptly processing irregular proteins to prevent pathological folding and aggregation. The proteasome system and lysosomal pathway are the two primary degradation routes within cells. Specifically, ubiquitin proteasomes break down short-lived and soluble misfolded proteins [5], while lysosomes degrade long-lived proteins, insoluble protein aggregates, and intracellular parasites [6, 7]. Beyond these, the ribonuclease (RNase) pathway and ClpCP proteases pathway also play significant roles in maintaining proteostasis. RNase-mediated modulators operate upstream, targeting RNAs that encode disease-related proteins, thereby influencing protein levels. The ClpCP protease system, found in some bacteria, acts as the functional counterpart to eukaryotic proteasomes [8].
These degradation pathways, excluding the RNA-targeting RNase pathway, form the foundation for targeted protein degradation (TPD). By leveraging the cell’s disposal mechanisms, TPD emerges as a promising therapeutic modality, offering access to a wide array of proteins. It requires only a target binder, or protein degrader, to execute its function. While conventional drugs like small molecule inhibitors and antibodies impact less than 20% of the proteome, TPD provides a unique approach to address the remaining, underexplored, and “undruggable” proteome with high precision. This allows for disease mitigation or cure by reducing harmful protein levels rather than modifying or inhibiting their functions. Furthermore, TPD can overcome certain cancer drug resistance mechanisms like gene mutation or overexpression [9].
The TPD concept was initially proposed in 1999 [10]. Crews and colleagues provided a more concrete proof-of-concept in 2001, establishing Arvinas in 2013, the first company solely dedicated to TPD. As TPD technologies rapidly advanced over the last two decades, various types of degraders have showcased the effectiveness, versatility, and transformative benefits of TPD. This review will briefly introduce cellular degradation pathways and corresponding TPD technologies. Table 1 summarizes representative TPD technologies based on their chemical components, target ranges, advantages, and potential drawbacks.
Table 1. Summary of representative TPD technologies related to different degradation pathways
| Pathway | TPD technologies | Target range | Composition | Advantages | Potential problems | Year | Refs |
|---|---|---|---|---|---|---|---|
| Proteasome | PROTAC | Intracellular | Small molecule/ biomacromolecule/ hybrid structure | Relatively high selectivity; Acceptable oral bioavailability; Clear degradation mechanism; Catalytic and sub-stoichiometric | Poor solubility for small-molecule PROTAC; Poor cell permeability; Poor PK properties; Limited target spectrum | 2001 | [11, 12] |
| Molecular glue | Intracellular | Small molecule | Acceptable oral bioavailability. | Difficult to design | 2010 | [13] | |
| SNIPER | Intracellular | Small molecule | Simultaneous degradation of POIs and IAPs; High specificity | E3 ligase IAPs dependently | 2010 | [14] | |
| HyT | Intracellular/ extracellular | Small molecule/ Small-molecule peptide conjugate | Some hydrophobic tags are independent of E3 ligases and ubiquitination; Wide range of potential targets; | Incomplete POIs degradation; Unclear degradation mechanism; Potential off-target effects | 2011 | [15] | |
| Trim-away | Intracellular | Antibody | High specificity; Rapid degradation speed | Need extra Trim21; Unable to recycle | 2017 | [16] | |
| Endosome- lysosome | LYTAC | Extracellular/ membrane proteins | Antibody | Degrade extracellular and membrane proteins; High controllability | Limited shuttle receptors; Potential immunogenicity; Non-catalytic; Low degradation efficiency | 2020 | [17, 18] |
| AbTAC | Membrane proteins | Bispecific antibody | Degrade membrane proteins; High specificity | Large molecular weight | 2021 | [19] | |
| GlueTAC | Extracellular/ membrane proteins | Nanobody-peptide conjugate | High specificity; Sufficient membrane permeability by a cell penetration peptide | Short half-life in vivo | 2021 | [20] | |
| Bispecific Aptamer Chimeras | Membrane proteins | Aptamer | Easy to design and prepare; Good stability | Low delivery efficacy; Short half-life in vivo | 2021 | [21] | |
| Sweeping antibody | Extracellular | Antibody | Allow recycling; | Required engineering for each target | 2013 | [22] | |
| Seldegs | IgG | Antigen-Fc fusion proteins | Degrade autoantibodies; Lower dose | Required engineering for each target; Antigen selection | 2017 | [23] | |
| Autophagy-lysosome | CMA-based degrader | Intracellular/ membrane proteins/aggregates | Chimeric polypeptides. | High specificity; High degradation efficacy | Low delivery efficacy; Low stability; Limited therapeutic effects; | 2014 | [24] |
| AUTAC | Intracellular/ damaged organelles | Small molecule-poly(A) oligonucleotide conjugate | A wide range of potential targets; Proteasome-independent | Low degradation speed; Potential off-target effects; Dependent on K63 ubiquitination; | 2019 | [25] | |
| ATTAC | Intracellular/ non-protein | Small molecule | A wide range of potential targets; Blood-brain barrier permeability; | Difficult to design | 2019 | [26, 27] | |
| AUTOTAC | Intracellular/ protein aggregates | Small molecule | Degrade protein aggregates | Low degradation speed | 2022 | [28] | |
| Ribonuclease | RIBOTAC | RNA | Small molecule/small molecule-poly(A) oligonucleotide conjugate | Expand targeted range to RNA; High degradation efficacy | Difficulties in finding specific ligands for targeting RNA | 2018 | [29, 30] |
| ClpCP proteases | BacPROTAC | Bacterial proteins | Small molecule/small molecule-peptide conjugate | Expand the targeted range to bacterial protein | Low efficiency | 2022 | [8] |
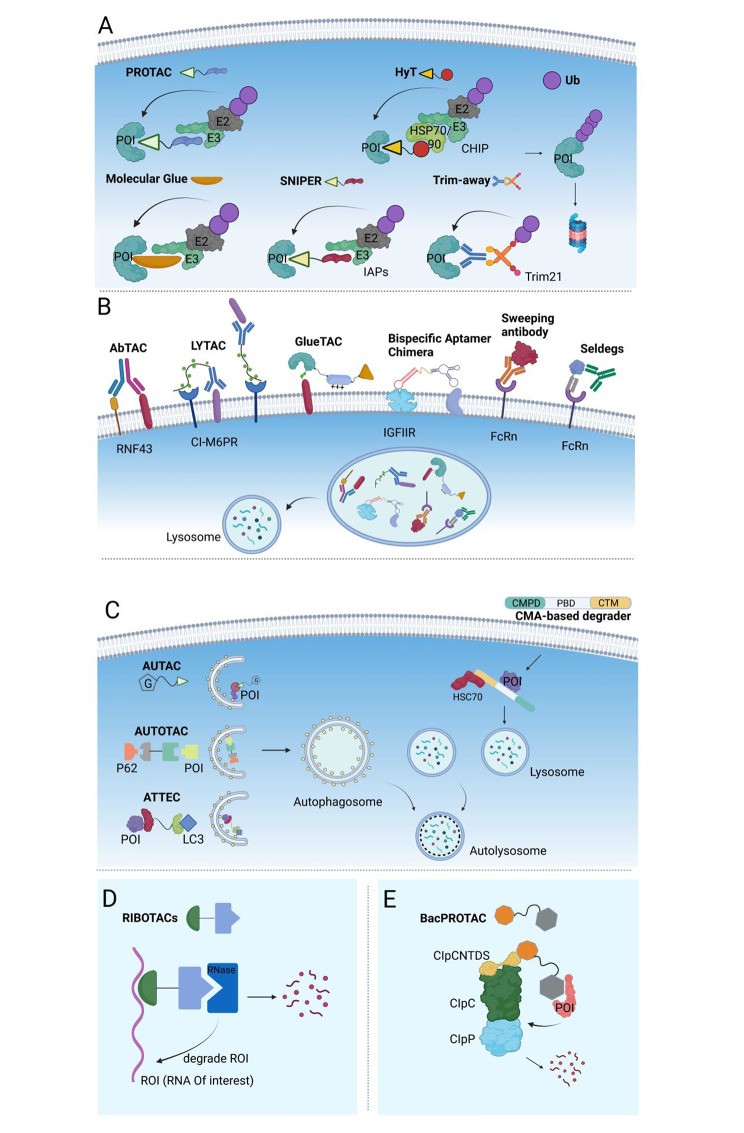
Open in a new tab
Table 1. An overview of various Targeted Protein Degradation (TPD) technologies, highlighting their mechanisms, target ranges, and compositions.
Abbreviations: POI, Protein of interest; IAPs, Inhibitor of apoptosis protein; HyT, Hydrophobic Tag; PROTAC, Proteolysis Targeting Chimeras; TPD, Targeted protein degradation; PK, Pharmacokinetics; SNIPER, Specific and Non-genetic IAP-dependent Protein Erasers; Trim21, Tripartite motif-containing protein 21; LYTAC, Lysosome-targeting chimeras; AbTAC, Antibody-based PROTAC; IgG, immunoglobulin G; CMA, Chaperone mediated autophagy; AUTAC, Autophagy-targeting chimera; ATTAC, Autophagy-tethering compounds; AUTOTAC, AUTOphagy-TArgeting Chimera; RIBOTAC, Ribonuclease targeting chimera
Ubiquitin-Proteasome Pathway: Precision Protein Removal
Dysfunctions within the ubiquitin system have been linked to the development of neurodegenerative conditions, Huntington’s disease, type II diabetes, and cancers [31–33]. As a crucial regulator of protein balance in eukaryotic cells, the ubiquitin-proteasome system degrades disease-related misfolded and abnormally aggregated proteins. This system offers significant potential for eliminating disordered proteins as a drug development strategy.
Ubiquitin (Ub), a highly conserved 76-amino acid protein, attaches to substrate proteins via lysine residues (primarily K63 and K48) [10] as a modifier in a process termed ubiquitination. Ubiquitination is a key posttranslational protein modification in eukaryotic cells. Beyond protein degradation, it plays a vital role in regulating diverse cellular processes, including protein transport, cell cycle, DNA repair, apoptosis, and signal transduction [34, 35]. The ubiquitin-proteasome system involves E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugation enzyme), and E3 (ubiquitin ligase) enzymes, along with the 26S proteasome [36, 37]. Ub is attached to substrate protein lysine residues through a sequential process involving these three enzymes. First, Ub forms a high-energy thioester bond with E1 in an ATP-dependent manner. The activated Ub is then transferred to the active site cysteine of E2. E3, working with E2, catalyzes the transfer of (poly)ubiquitin to the protein targeted for degradation [38, 39]. Finally, the 26S proteasome recognizes the polyubiquitinated protein and degrades it into small peptides.
Due to their capacity to facilitate substrate protein ubiquitination and drive proteasomal degradation, E3 ligases are a primary focus in TPD research. In 2001, Crews’ group [11] developed the first proteolysis-targeting chimera (PROTAC) using the Skp1-Cullin-F-box (SCF) complex to target methionine aminopeptidase-2 (METAP2) for degradation. By designing E3 ligands and linking them to target protein conjugates, various ubiquitin proteasome-based TPD strategies have been developed to degrade specific proteins [40–42]. Among these, PROTAC, hydrophobic tags (HyT) [15], and specific and non-genetic inhibitors of apoptosis protein-dependent protein erosive agents (SNIPER) [14] are bispecific chimeric molecules that simultaneously bind proteins of interest (POIs) and E3 ligases, enabling POI ubiquitination and subsequent proteasomal degradation. Unlike these degraders, molecular glues are small chemicals that bind either the ligase (mostly) or the POI to induce proteasomal degradation [13, 43]. The TRIM-away system utilizes tripartite motif-containing protein 21 (TRIM21, an E3 ligase recognizing the Fc fragment of an antibody) to target antibody-POI complexes or antibody-bound pathogens to the proteasome for degradation [16] (Fig. 1A).
Fig. 1.
Open in a new tab
Fig. 1. Visual representation of five distinct Targeted Degradation Pathways: Proteasome, Endosome-lysosome, Autophagy-lysosome, Ribonuclease, and ClpCP proteases.
Targeted degradation via five distinct degradation pathways. (A) Proteasome pathway. Molecule glue is a monovalent small molecule degrader that employs a single interaction with the POI or an E3 ligase, whereas PROTAC, hydrophobic tags (HyT), and SNIPER are chimeric molecules that simultaneously bind to the POI and the E3 ubiquitin ligase. These degraders enable POI ubiquitination and subsequent proteasomal degradation. TRIM-away system consists of an antibody and TRIM21. TRIM21, an E3 ligase recognizing the Fc fragment of an antibody, can facilitate the antibody-POI complex or antibody-bound pathogens to the proteasome for degradation. (B) Endosome-lysosome pathway. AbTAC, LYTAC, bispecific aptamer chimeras, GlueTAC, sweeping antibody, and Seldegs develop an ‘outside-in’ strategy to shutter extracellular/membrane POIs to the endosome and undergo lysosomal degradation. (C) Autophagy-lysosome pathway. AUTAC, ATTEC, and AUTOTAC, also chimeric molecules, link the intracellular substrate and adaptor proteins (e.g. LC3, p62) or autophagosome, which was fused with lysosome and processed to degradation. CMA-based degraders degrade membrane/intracellular proteins by harnessing chaperone-mediated autophagy (CMA), rather than macroautophagy. (D) Ribonuclease pathway. RIBOTAC is a targeted RNA degradation technology, which recruits a nuclease to a specific RNA and triggers its collapse. (E) ClpCP proteases pathway (bacterial degradation machinery). BacPROTAC tethers the target bacterial protein to the ClpC:ClpP protease and then primes the neo-substrates for degradation. The figure was created in BioRender.com
Lysosomal Degradation Pathway: Expanding the Scope of TPD
While proteasome-mediated TPD is a potent tool for modulating undruggable protein targets, its application is largely confined to soluble intracellular proteins [16]. Like the ubiquitin-proteasome system, the lysosomal system is essential for maintaining protein homeostasis and the integrity of both intra- and extracellular environments [44]. Compared to the proteasomal system, lysosomes can degrade a broader range of substrates, including soluble proteins, aggregated proteins, non-proteinous components, and even organelles. Lysosomal degradation has emerged as a promising modality for TPD technology and an alternative to ubiquitin-proteasome system-based degradation techniques.
In recent years, TPD methods employing the lysosomal degradation pathway, such as antibody-based PROTAC (AbTAC), lysosome-targeting chimeras (LYTAC), GlueTAC, bispecific aptamer chimeras, AUtophagy-targeting chimeras (AUTAC), AUTOphagy-targeting chimeras (AUTOTAC), and autophagy-tethering compounds (ATTEC) have been developed [10, 45]. Endosome-lysosome and autophagy-lysosome are two lysosomal degradation pathways commonly utilized in TPD.
Endocytosis is a fundamental process where the plasma membrane folds inward, engulfing external materials into vesicles. These vesicles then undergo a series of steps to become endosomes and eventually fuse with lysosomes to digest their contents [46]. Fluid-phase endocytic uptake is associated with clathrin-mediated endocytosis, caveolin-mediated endocytosis, clathrin/caveolae-independent endocytosis, and macropinocytosis [47]. Phagocytosis, a specialized form of endocytosis, allows cells to transport harmful substances like bacteria, viruses, and pathogens to lysosomes for degradation, protecting cells from external threats [48].
Ongoing research into the endocytic lysosomal pathway has significantly expanded the TPD scope from intracellular targets to extracellular and membrane targets through new TPD strategies. These include AbTAC (a bispecific antibody targeting a transmembrane E3 ligase and a membrane-related protein) [19], LYTAC (an antibody/small molecule targeting a POI and a lysosomal shuttle receptor) [17, 18], GlueTAC (a covalent nanobody fused to a cell-penetrating peptide/lysosome sorting sequence (CPP-LSS)) [20], bispecific aptamer chimeras (a bispecific aptamer chimera binding to a lysosomal shuttle receptor and a transmembrane protein) [21], sweeping antibody [22] and Seldeg [23] (both engineered antibodies hijacking Fc receptors) (Fig. 1B).
Besides the endosome-lysosome pathway, the autophagy-lysosome pathway provides another route in TPD. Autophagy, a highly conserved degradation mechanism in yeast and mammals, is crucial for maintaining intracellular homeostasis and normal metabolic function. It breaks down dysfunctional intracellular proteins and damaged organelles to generate recyclable nutrients like amino acids and lipids [49, 50]. Macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) are three specific forms of the autophagic lysosomal pathway [51].
Macroautophagy begins with a detached membrane structure, the phagophore, derived from a phospholipid bilayer containing lipidated LC3. This phagophore expands to engulf autophagic substrates, including proteins and organelles, sequestering them in a double-membrane vesicle called the autophagosome. Cargo degradation occurs after these autophagosomes fuse with lysosomes [52]. Microautophagy is a non-selective phagocytic process where the lysosomal membrane directly engulfs cytoplasmic cargo, which is then degraded by hydrolases [53]. During CMA, proteins with specific motifs (KFERQ) are selected by chaperones, targeted to lysosomes, and directly translocated into the lysosomal lumen for clearance [54].
AUTAC, comprising an S-guanine tag and a warhead for intracellular POIs via a flexible linker, was the first degrader developed to target the autophagy machinery. AUTAC recruits autophagosomes through K63 polyubiquitination and directs substrates for selective autophagy [25] (Fig. 1C). Unlike AUTAC, which uses selective autophagy (xenophagy), ATTEC and AUTOTAC directly engage the autophagy pathway. LC3 (Atg8) and p62, widely used autophagy markers, are linked to autophagy initiation and have been used to develop ATTEC and AUTOTAC. An ATTEC molecule simultaneously binds LC3 and a POI [26], while an AUTOTAC molecule binds p62 and a POI [28]. These chimeric structures recruit autophagosomes, leading to autophagy-mediated degradation (Fig. 1C). CMA-based degraders [24], typically peptide-based, contain a cell membrane penetrating domain (CMPD), a protein binding domain (PBD), and a CMA sorting signal. This fused peptide drives protein clearance through the CMA pathway (Fig. 1C).
Alternative Targeted Degradation Pathways: RNA and Bacterial Protein Degradation
RNase Pathway: Targeting RNA for Degradation
The ENCODE project has revealed that while only 1–2% of the human genome codes for proteins, at least 76% is transcribed into RNA [55]. Non-coding RNAs, including microRNA, lncRNA, and intron RNA, play crucial roles in regulating gene and protein expression. As key regulators of biological functions, RNA production and removal are tightly controlled. RNases are nucleases that naturally regulate RNA lifespan. Exploiting RNases offers a promising approach to regulate RNA fates via chimeric structures similar to PROTACs.
Disney’s group has made significant contributions to expanding TPD from proteins to RNA. Using their Inforna method to design small molecules targeting RNA [56], Disney’s group [29] developed the first RNase targeting chimera (RIBOTAC). This chimera links a short 2’-5’ A4 oligonucleotide targeting RNase L with a small molecule that recruits the primary transcript of microRNA-96 (pri-miR-96) (Fig. 1D). Importantly, the RIBOTAC degrader not only recruits inactive RNase L to the target RNA but also activates its catalytic activity upon conjugation. In response to the COVID-19 pandemic, Disney’s group [30] designed RIBOTACs to destroy the frameshifting element within the SARS-CoV-2 RNA genome in 2020. These RNA degraders specifically bind to the revised attenuator hairpin structure of the viral RNA, suggesting a potential tool for targeting the SARS-CoV-2 RNA genome.
Compared to other RNA silencing technologies like antisense oligonucleotides and siRNA, RIBOTAC offers advantages, including catalytic properties and improved bioavailability [29].
ClpCP Protease Pathway: Bacterial Protein Degradation
PROTACs eliminate proteins by engaging the eukaryotic ubiquitin-proteasome system. However, this technology is limited to eukaryotes and cannot be applied to bacteria, which lack a ubiquitination system. While ubiquitin is unique to eukaryotic cells, some prokaryotic cells have similar degradation markers. A short phosphorylated arginine residue fragment (pArg) acts as a hydrolysis tag recognized by the bacterial ClpC:ClpP (ClpCP) protease system, the functional equivalent of the eukaryotic proteasome in gram-positive bacteria and mycobacteria [57]. Compared to the eukaryotic proteasome that recognizes polyubiquitin signals, the ClpCP protease recognition mechanism is simpler: a pArg tag is attached to the target protein and is recognized by ClpCP protease as a degradation signal [57].
Recently, Morreale et al. [8] developed the first bacterial PROTACs (BacPROTACs) that redirect the ClpCP protease to degrade neo-substrates, expanding TPD application to bacteria and offering a novel platform for antibiotic discovery (Fig. 1E). A pArg group or pArg-like cyclic peptides were chosen as ligands targeting the ClpCP protease. Structural studies indicate that BacPROTAC protease ligands not only serve as targeting moieties but also convert ClpC into active, higher-order oligomers with ClpP. Designed BacPROTACs showed high affinity for the protease and efficient degradation activity in vivo.
Development and Disadvantages of Small Molecule PROTACs: Lessons for BioTPD
Crews et al. published the first report on PROTAC technology based on SCF in 2001 [11]. To target a protein to the SCF complex, a METAP2 ligand (ovalicin) was linked to a βTRCP E3 ligase ligand (I kappa Bα (IκBα) phosphopeptide) via a linker. This PROTAC promoted METAP2 ubiquitination and destruction in a cell-free system, providing the first proof-of-concept for PROTAC-mediated target protein degradation in vitro.
In 2003, Crews et al. again used this approach to degrade estrogen receptor (ER) and androgen receptor (AR) [12]. Microinjection of a dihydrotestosterone-IκBα phosphopeptide PROTAC molecule into 293GFP-AR cells induced significant GFP-AR degradation, demonstrating that PROTACs are not limited to extracellular space and can trigger protein degradation in cells via the proteasomal pathway.
Early PROTAC construction used IκBα phosphopeptide as a binder for β-TRCP E3 ligase. Researchers also identified short peptides binding to von Hippel-Lindau (VHL), an E3 ubiquitin ligase targeting tumor-associated transcription factor hypoxia-inducible factor 1 (HIF1) [58, 59]. Zhang lab [60] created Fumagillo/estradiol-octapeptide (a HIF1 ligand) PROTACs targeting METAP2 and ER, observing target protein ubiquitination after cell treatment. Schneekloth et al. [61] developed PROTACs containing a short peptide (heptapeptide) as a VHL ligand tethered to AP21998 or DHT, targeting FKBP12 or AR, respectively.
These early PROTACs are now termed ‘bioPROTACs’ as they incorporate peptide ligands for E3 targeting [62]. Unmodified peptide chain PROTACs face challenges entering cells due to high polarity and poor permeability [63]. Low peptide stability also impacts degradation effectiveness [64]. Researchers sought small molecules with peptide ligand functions to bind E3 ligases, improving PROTAC pharmacokinetic properties and stability.
In 2008, Crews lab [65] created the first fully small-molecule PROTAC using mouse double minute 2 homolog (MDM2), an E3 ligase targeting p53. The PROTAC comprised SARM, a small-molecule AR ligand [66], and nutlin, a small-molecule MDM2 ligand [67]. Significant AR degradation was seen in cells treated with SARM-nutlin PROTAC. This cell-permeable small molecule PROTAC spurred the development of numerous similar compounds.
Inhibitor of apoptosis protein 1 (cIAP1) is the second E3 ligase, after MDM2, used in small molecule PROTAC construction. Sekine et al. [68] reported ME-BS, a small molecule selectively downregulating cIAP1 by interacting with its baculovirus IAP repeat 3 (BIR3) domain, promoting ubiquitination and self-degradation. Various ME-BS-based small-molecule PROTACs emerged, like ATRA-MEBS [69] and 4-OHT-MEBS [70].
Small molecule VHL-recruiting PROTACs were also developed by replacing the HIF1 peptide with high-affinity small-molecule ligands [71, 72]. Subsequently, VHL-based small molecule PROTACs were developed to target and degrade various proteins, including receptor-interacting serine/threonine kinase 2 (RIPK2) [73], BCR-ABL [74], TANK-binding kinase 1 (TBK1) [75], epidermal growth factor receptor (EGFR) [76], tripartite motif containing 24 (TRIM24) [77], and bromodomain-containing 4 (BRD4) [78–80].
Small molecule PROTACs based on E3 ubiquitin ligases like SCFβ−TRCP, cereblon, RING finger protein 4 (RNF4), RING finger protein 114 (RNF114), and Kelch-1ike ECH- associated protein l (KEAP1) have also been developed [81–85]. Since the first small-molecule PROTAC, the technology has expanded from academia to industry, with pharmaceutical companies developing PROTAC pipelines for clinical translation, driven by favorable cell permeability, stability, degradation efficiency, and longer action duration [86–89].
However, small-molecule PROTACs have disadvantages: (1) reliance on target protein binding pockets [11]; (2) dependence on the proteasome system [90]; (3) off-target effects and potential adverse reactions [91, 92]; and (4) the Hook effect at saturating doses, reducing degradation efficiency [93].
Advances in bioTPD: Expanding Therapeutic Horizons
Biological TPD (bioTPD), including peptide-based PROTACs and non-small molecule TPD technologies using nucleic acids or proteins [10, 45, 94], offers advantages over small-molecule TPD. These include: (1) antibodies and peptides can specifically bind undruggable proteins, unaffected by target protein binding pockets, facilitating bioTPD construction [95]; (2) protein- and peptide-based bioTPD are easier to design and synthesize, with better safety and lower toxicity [96]; (3) protein/peptide ligands can recognize mutated targets [97], reducing off-target effects; (4) LYTAC, GlueTAC, nucleic acid PROTAC, and other lysosomal pathway-dependent TPDs can degrade membrane and extracellular proteins [10]; and (5) peptide or antibody (fragment) ligand affinity and specificity are often higher than small-molecule compounds, potentially leading to higher bioTPD efficiency and selectivity. While direct comparisons of degradation speed/efficiency between bioTPD and small-molecule TPD are limited, further research is warranted. BioTPD expands the PROTAC toolbox, providing new protein degradation options beyond small molecules.
This review categorizes bioTPD into peptide-, fusion protein-, antibody (fragments)-, and nucleic acid-based subgroups, focusing on their development, components, mechanisms, features, and potential applications, along with their limitations.
Peptide-Based bioTPD: Early Innovations and Continued Relevance
Peptide-based bioTPD, especially peptide PROTACs, represents the earliest TPD technology. Despite pharmacokinetic limitations, peptide degraders remain complementary to small-molecule degraders. Peptide-based bioTPD degraders include whole peptides and hybrids combining peptide ligands and small-molecule warheads. Before small-molecule ligands for VHL, cereblon, and Keap1 were discovered, peptide ligands were widely used for E3 targeting. We now describe peptide degraders based on different E3 ligases and lysosomal adaptor proteins (Table 2).
Table 2. Representative example of peptide-based bioTPD
| Pathway | Adaptor | Key sequences | POI | POI ligands | Refs |
|---|---|---|---|---|---|
| Proteasome | VHL | MLAP(OH)YIPM | METAP2 ER | Fumagillo Estradiol | [60] |
| LAP(OH)YI | AR ER CREPT | Dihydrotestosterone Estradiol VRALKQKYEELKKEKESLVDK | [100, 104] | ||
| ALAPYIP | Akt FRS2α PI3K Tau | Recognition peptide for Akt2 (P-ser474) IENPQYFSDA GPGGDYAAMGACPASEQGYEEMRA YQYQDATADEQG | [109, 110, 114] | ||
| β-TRCP | DRHDS(P)GLDS(P)M | METAP2 | Ovalicin | [11] | |
| KEAP1 | LDPETGEYL | Tau | YQYQDATADEQG | [118] | |
| / | RRRG | α-synuclein | GVLYVGSKTR | [136] | |
| Lysosome | / / / | KFERQKILDQRFFE MDFSGLSLIKLKKQ cyclic RGDyK | DAPK1 α-synuclein PD-L1 PD-L1 | A fragment of the GluN2B subunit A short peptide of β-synuclein DKEMAATSAAIEDAVRRIEDMMNQ BMS-8 | [24] [131] [132] |
Open in a new tab
Table 2. Examples of peptide-based bioTPD approaches, showcasing various adaptor proteins, targeting sequences, and proteins of interest.
Abbreviations: VHL, Von Hippel-Lindau; METAP2, Methionine aminopeptidase 2; ER, Estrogen receptor; AR, Androgen receptor; CREPT, Cell cycle-related and expression-elevated protein in tumor; Akt, Serine/threonine-protein kinase AKT; FRS2α, Factor receptor substrate 2α; PI3K, Phosphatidylinositol-3-kinase; Tau, microtubule-associated protein; α-synuclein, alpha-synuclein; DAPK1, Death associated protein kinase 1; PD-L1, programmed cell death ligand 1
VHL-Dependent Peptide bioTPD: Fine-Tuning Degradation
(1) MLAP(OH)YIPM: Precision Targeting of ER and METAP2
Zhang et al. [60] created a small molecule protein hydrolysis inducer (SMPI) targeting ER and METAP2 by combining estradiol/fumagillo with MLAP(OH)YIPM, a short HIF-derived peptide [98]. These chimeras significantly increased METAP2 ubiquitination and ER degradation in lung cancer A549 and breast cancer MCF-7 cells in a time-dependent manner. Rescue experiments showed that replacing ProOH with Ala eliminated protein degradation capability, indicating hydroxylated proline’s necessity for VHL binding [85] and its importance in VHL-binding peptides.
Building on this, Zhang et al. [60] modified MLAP(OH)YIPM to a pentapeptide, removing flanking amino acids while keeping ProOH. PROTAC (E2-penta), combining this pentapeptide with estradiol, also effectively increased ER degradation. This simplification of the peptide chain while retaining the crucial ProOH group not only streamlines PROTAC manufacturing but also provides a basis for developing peptide mimics.
(2) LAP(OH)YI: Efficient ER Degradation and Tissue-Level Impact
In 2004, Zhang et al. [60] detailed that short pentapeptide structures with ProOH bind VHL. Bargagna-Mohan et al. [99] tested ProOH-based-domain-estradiol PROTACs with varying peptide chain lengths (5–8 amino acids) to degrade ER at the tissue level using a 3D endothelial cell germination assay (3D-ECSA). PROTACs built with LAP(OH)YI showed more efficient ER degradation than octapeptide PROTACs.
LAP(OH)YI was further used by Rodriguez-Gonzalez et al. [100] to create PROTAC-A and PROTAC-B with dihydrotestosterone/estradiol to suppress AR and ER in prostate and breast cancer cells, respectively. After 72 hours, both PROTAC-A and PROTAC-B inhibited cell proliferation. PROTAC-B reduced cyclin D1 and progesterone receptor (PR) expression and blocked downstream signaling in MCF-7 and T47D cells. Rodriguez-Gonzalez et al. [101] also created PROTAC-AA by adding a polyarginine tail and two glycine residues to PROTAC-A to enhance cell penetration. PROTAC-AA showed at least a five-fold IC50 decrease compared to PROTAC-A, demonstrating polyarginine tails as a powerful strategy for improving PROTAC permeability and solubility.
Cell cycle-related and expression-elevated protein in tumor (CREPT, or RPRD1B) is elevated in various cancers. CREPT, an RNA polymerase II-associated protein, promotes cyclin D1 transcription by inducing chromatin loop formation and activating transcription in response to Wnt signaling [102, 103]. The leucine-zipper-like motif, a typical alpha-helix motif for protein homodimerization, was used as the CREPT binding component [104, 105]. Speltz et al. [106] constructed a PROTAC linking this sequence to LAP(OH)YI, with a membrane penetrating sequence (KRRRR) at the C-terminus. CREPT degradation was observed after treating Panc-1 cells with this PROTAC. Dimerization sequences offer a novel approach to finding target protein binders for PROTAC development, especially for proteins not bound by small molecules.
(3) ALAPYIP: Versatile VHL-Binding Peptide for Diverse Targets
ALAPYIP, another natural VHL-binding short peptide from HIF1-VHL interaction [58], was first used by Schneekloth et al. [61] in PROTAC construction by linking a ligand for FK506 binding protein (FKBP12) and AR. These PROTACs efficiently drove proteasomal degradation of both targets.
Protein kinase B (PKB or AKT), a serine/threonine protein kinase involved in cellular metabolism, apoptosis, and cell growth, is aberrantly signaled in multiple cancers and diabetes [107], making it a challenging drug target. Using protein-catalyzed capture agents (PCCs) [108], a high-affinity PCC for Akt was identified and functionalized with a VHL-binder (ALAPYIP) and HIV TAT, resulting in remarkable cell permeability and high degradation efficacy (over 90%) after 4 hours [109].
Hines et al. [110] developed phosphorylation-dependent PROTAC (phosphoPROTAC) techniques for selective degradation of proteins in activated kinase-signaling pathways. Phosphorylation motifs of tropomyosin receptor kinase A (TrkA) and erythroblastosis oncogene B3 (ErbB3) were chosen as ligands to recruit fibroblast growth factor receptor substrate 2α (FRS2α) and phosphatidylinositol-3-kinase (PI3K), respectively [111, 112]. Cell-permeable phosphoPROTACs were created by joining the VHL recognition peptide ALAPYIP with TAT to these phosphorylation motifs, which become phosphorylated upon receptor tyrosine kinase (RTK) activation, binding to FRS2α or PI3K, and triggering proteasomal destruction. PhosphoPROTAC’s advantage is cell-type-selective degradation based on tyrosine kinase pathway states. It is also less prone to drug resistance, as conditional destruction relies on misregulated kinase activity, reducing selective pressure for target kinase mutations, unlike small molecule inhibitors that broadly inhibit kinases.
To specifically degrade β-catenin, a multifunctional protein in cell adhesion and Wnt signaling, Liao et al. [113] first synthesized two β-catenin-specific stapled helical peptides (SAHPA1 and xStAx) with improved membrane permeability and stability using peptide stapling chemistry. They then created PROTAC (xStAx-VHLL), a potent β-catenin degrader, combining xStAx with the VHL ligand ALAPYIP. This study highlights peptide-based PROTACs as a new drug class to address Wnt/β-catenin signaling-related diseases via β-catenin degradation.
A PROTAC containing the Tau recognition motif YQYQDATADEQG, a CPP, and the VHL binding motif ALAPYIP was also reported to target Tau protein degradation in the mouse brain [114], marking a new era in central nervous system disease treatment with multifunctional peptides.
SCFβ−TRCP-Dependent Peptide (DRHDS(P)GLDS(P)M) bioTPD: Targeting METAP2
IκBα, a negative regulator of nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB), binds SCFβ−TRCP upon inflammatory stimuli. IκBα recruitment to SCFβ−TRCP depends on a 10-amino acid peptide within IκBα (DRHDSGLDSM) with two serines that can be phosphorylated in response to inflammatory signals. SCFβ−TRCP binds the phosphorylated sequence, triggering ubiquitination and destruction [115]. Sakamoto et al. [11] constructed PROTAC (ovalicin-DRHDS(P)GLDS(P)M) using this phosphorylation sequence and demonstrated that it promoted METAP2 ubiquitination in vitro. Later studies [12] constructed similar PROTACs targeting ER and AR, observing target protein degradation in cells via microinjection.
KEAP1-Dependent Peptide (LDPETGEYL) bioTPD: Degrading Tau Protein
Keap1 is a substrate adaptor protein for the Cullin3 (CUL3)/Ring-Box1 (Rbx1) E3 ubiquitin ligase complex. Transcription factor NF-E2-related factor 2 (Nrf2) is a known Keap1-CUL3 substrate, with the Nrf2-Keap1 pathway playing a major role in cellular defense against oxidative stress [116]. Lu et al. [117] identified a short peptide (LDPETGEYL) that could inhibit Keap1-Nrf2 interaction and strongly bind Keap1. They created a full peptide PROTAC [118] assembling this Keap1 recognition domain with YQQYQDATADEQG (a Tau-targeting peptide) and poly-D-arginine (CPP). Applying this PROTAC to Tau-overexpressing cells (SH-SY5Y, N2a, and PC-12 cells) resulted in proteasome-dependent downregulation of intracellular Tau.
Proteasome-Dependent Peptide (RRRG) bioTPD: Targeting α-synuclein
Bonger et al. [119] developed a method for protein degradation using small molecule Shield-1. The POI was genetically fused with a ligand-induced degradation (LID) domain, creating a stable product. Shield-1 addition rapidly destabilized the LID domain and induced target degradation. The LID domain contains a 19-amino acid degron appended to the C-terminus of FK506- and rapamycin-binding protein (FKBP). Truncation experiments revealed that only a 4-amino acid sequence (RRRG) from the 19-amino acid degron was needed for degradation, which proteasome inhibitor MG132 could completely prevent. However, the exact degradation mechanism remains unknown.
Lewy bodies (LBs) in surviving neurons are a hallmark of PD pathogenesis. α-synuclein (α-syn) is the main LB component, a small 140-amino acid protein highlighted as a major driver of PD pathogenesis [120]. Lysosomal α-syn degradation by peptide-based TPD has been shown [24]. If autophagy-lysosomal function is compromised in disease progression, proteasomal α-syn clearance becomes an alternative. Qu et al. [121] used the RRRG peptide to create a bifunctional degrader consisting of an α-syn binding sequence, RRRG (proteasome targeting sequence), and TAT, inducing intracellular α-syn degradation in a concentration- and time-dependent manner [119, 122, 123].
Lysosome-Dependent Peptide bioTPD: CMA and RGD-Integrin Targeting
(1) KFERQ/HSC70: CMA-Mediated Degradation
CMA is a type of autophagy specific for substrate proteins containing a pentapeptide motif (KFERQ). Heat shock cognate protein 70 (HSC70) recognizes KFERQ, forming a substrate/chaperone complex. When near the lysosome, this complex binds to the lysosome-associated membrane protein type 2A (LAMP2A), causing LAMP2A multimerization and substrate protein destruction [124, 125]. KFERQKILDQRFFE, created by Fan et al. [24], links KFERQ with two other CMA-targeting motifs (CTM), QKILD and QRFFE. They showed that non-CMA substrate proteins fused with this sequence could be directed to CMA lysosomal degradation. They designed a CMA-based degrader with a cell membrane penetrating domain (CMPD), a target protein binding domain (PBD), and a CMA targeting domain (CTM). Fan et al. designed HA-GluN2Bct-CTM, where GluN2Bct only binds the active, but not inactive, form of death-associated protein kinase 1 (DAPK1) [126]. Coexpression of HA-GluN2Bct-CTM with cDAPK1 (active DAPK1) in HEK cells resulted in cDAPK1 degradation. TAT-GluN2Bct-CTM, cell-permeable via TAT, was created. Co-treating HEK cells expressing WT DAPK1 with TAT-GluN2Bct-CTM and NMDA (a DAPK1 activator) showed NMDA stimulation promoted WT DAPK1 degradation. To test degradation efficacy on other proteins, TAT-βsyn-CTM and TAT-GluN2B9c-CTM were created for targeting α-syn and postsynaptic density protein 95 (PSD-95). Both peptides at 25 µM degraded corresponding target proteins in neuronal cells.
Cyclin-dependent kinase 5 (CDK5), a proline-directed serine/threonine kinase, overactivation of which is linked to neuronal cell death in stroke [127]. Targeted abnormal CDK5 degradation protects injured neurons. Zhou et al. [128] constructed a CDK5-targeted degradation peptide (TAT-CDK5-CTM) using KFERQ and treated cortical neurons (OGD induction) with 5 µM TAT-CDK5-CTM. Significant CDK5 degradation and reversal of OGD-induced damage were observed.
(2) MDFSGLSLIKLKKQ/Di-leucine Sorting Signals: Targeting PD-L1
Programmed death ligand 1 (PD-L1), a transmembrane protein overexpressed in many cancers, is linked to immune escape, making it a popular oncology target [129]. Using the OncoBinder approach [130], Wang et al. [131] found that huntingtin-interacting protein 1-related protein (HIP1R) interacts with and negatively regulates PD-L1, targeting PD-L1 to lysosomal degradation via a lysosomal sorting signal in HIP1R (966–979). Inspired by Fan et al. [24], they combined this sequence with the PD-L1 binding sequence in HIP1R (784–807) to form a fusion peptide, PD-LYSO. Significant PD-L1 degradation was observed after overexpressing PD-LYSO in cells. The lysosomal sorting effect of HIP1R (966–979) is due to a di-leucine sorting signal sorting cargos to the lysosome, not CMA.
(3) RGD-Integrin-Mediated TPD: Targeting Extracellular and Membrane Proteins
Recently, Fang et al. [132] demonstrated RGD-integrin-mediated TPD. They created a bifunctional compound with a POI-binding ligand and a cyclic RGD peptide as the integrin-binding ligand. The resulting degrader induces integrin- and lysosome-dependent internalization and degradation of extracellular (NeutrAvidin protein, apolipoprotein E4) or cell membrane proteins (PD-L1). Since αvβ3 integrin is often overexpressed in cancers, this strategy is attractive for targeted degradation of cancer-relevant proteins. It may be extended to other cell-surface receptors like the transferrin receptor [133] and folate receptor [134] via receptor-mediated endocytosis and lysosomal degradation.
ClpCP Protease-Based Peptide bioTPD: Targeting Bacterial Proteins
ClpCP protease, a protein-degrading enzyme recognizing pArg as a degradation tag, is the basis of BacPROTAC technology. Morreale et al. [8] designed BacPROTAC degraders using bacterial ClpCP. BacPROTAC-1, a chimeric small-molecule degrader for monomeric streptavidin (mSA), links a pArg derivative (ClpCP ligand) to biotin (a high-affinity mSA ligand) via a linker. BacPROTAC-1 has high affinity for mSA (KD = 3.9 µM) and ClpCP (KD = 2.8 µM), inducing degradation of mSA and three mSA fusion proteins. To address pArg’s instability and poor pharmacokinetics, Morreale et al. replaced pArg with Cyclomarin A (CymA), a cyclic peptide antibiotic targeting mycobacterial ClpC1 [135] with pArg-like function. CymA was modified to obtain sCym-1 (KD = 0.81 µM), a high-affinity ClpC1 ligand. sCym-1, as a ClpC1 protease ligand, was linked to JQ1 (an inhibitor of bromodomain-1 (BD1) of BRDT) to form BacPROTAC-3. This degrader induced BRDTBD1 degradation in a concentration-dependent manner both outside and inside bacteria. BacPROTACs show that bacterial proteins can be selectively degraded via targeted protease pathways. Identifying bacterial protein ligands and linking them to CymA/CymA modifiers to create ClpCP protease-based BacPROTACs is a feasible strategy for microbial infections.
Fusion Protein-Based bioTPD: Genetically Engineered Degradation
Fusion proteins, complex proteins fusing a target protein binding sequence with a full-length or truncated E3 ligase, differ from traditional PROTACs with target protein, linker, and E3 ligase junctions. Fusion protein degrader developers genetically engineer E3 ligases to change substrate specificity. The hook effect, intrinsic to TPD molecules needing ternary complex formation, is largely avoided as fusion protein-based bioTPD already contains the E3 ligase module, eliminating ternary complex formation.
Over 600 human E3 ligases are identified, but only ~10 are used in classical PROTACs. Fusion protein-based bioTPD has advanced TPD strategies and applications. The three main E3 ubiquitin ligase types are Really Interesting New Gene (RING), Homologous to the E6-AP Carboxyl Terminus (HECT), and RING-between-RING (RBR) E3s, with RING E3s being most abundant [39]. Reported fusion protein-based degraders mainly hijack RING-type E3 ligases, including Hsc70-interacting protein (CHIP), Speckle-type POZ protein (SPOP) VHL, RNF4, and SCFβ−TRCP. This section describes fusion protein-based bioTPD development with different E3 ligases as backbones (Fig. 2).
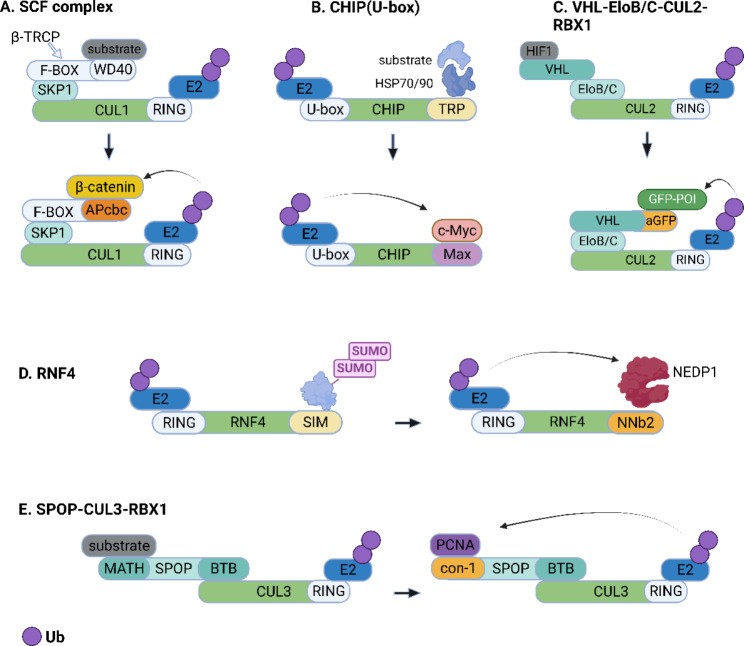
Fig. 2.
Open in a new tab
Fig. 2. Diagram illustrating various fusion protein-based bioTPD strategies, showcasing different E3 ligases and targeting motifs.
Schematic representation of fusion protein-based bioTPD. (A) A β-catenin-binding motif (APcbc) replaces the natural substrate-binding domain of β-TRCP (WD40, etc.) to form a fusion protein for targeted degradation of β-catenin. (B) Max (a binding partner of c-Myc) is linked to the U-box of CHIP to form an artificial ubiquitin ligase for targeted destruction of c-Myc. (C) An affinity-directed protein missile (AdPROM) system harbors an anti-GFP nanobody (aGFP) that is fused to VHL to recruit any GFP-tagged protein (GFP-POI) to the CUL2 E3 ligase machinery. (D) The antibody RING-mediated destruction (ARMeD) system is mediated by a NEDP1-targeting nanobody (NNb2) fused to the RING domain of ubiquitin E3 ligase RNF4 for targeted destruction of NEDP1 via the ubiquitin-proteasome system. (E) A high-affinity peptide for PCNA (con-1) replaces the substrate-binding MATH domain of the E3 adaptor SPOP, which enable the ubiquitin tagging of PCNA by the CUL3-based Cullin-RING ligase complex. The figure was created in BioRender.com
SKP1-CUL1-F-box-RBX1-Based Fusion Protein: Early PROTAC Mimicry
Since first PROTACs [11], PROTAC structure was seen as fixed: POI binding component, linker, and E3 ligase binding component. However, in 2000, Zhou et al. [137] constructed a PROTAC-like fusion protein, also tethering target protein to an E3 ligase, prompting fusion protein-based bioTPD development.
SCFβ−TRCP, a common E3 ligase, comprises Rbx1 (RING domain), Cullin1 scaffold, and F-box protein/SKP1 complex [138], with F-box protein being key. F-box protein has two domains: F-box domain binding SKP1 and substrate recognition domain (WD40 or leucine-rich repeat sequences for substrate protein binding). Yeast-derived F-box protein (Cdc4p) was modified by attaching its terminus to the retinoblastoma protein pRB-binding fragment (E7N), forming the Cdc4pF/WD-E7N complex. pRB degradation was seen in yeast cells expressing pRB treated with this engineered protein, with a half-life <60 min. Similar modification on human F-box protein β-TRCP showed similar protein degradation effects. Su et al. [139] similarly modified F-box protein β-TRCP to selectively eradicate pathogenic β-catenin (Fig. 2A). Analyzing β-catenin interacting motifs, a 15-amino acid peptide (APCbc) strongly binding β-catenin was identified. F3APCbc4 is a fusion protein with four APCbc repeat units linked to the F-box structural domain by a linker. F3APCbc4 treatment reduced β-catenin and attenuated downstream signal Myc.
To better study intracellular protein function, Caussinus et al. [140] developed deGradFP, a GFP fusion protein degradation method, using Slmb, a Drosophila melanogaster-derived F-box protein. NSlmb-vhhGFP4, formed by linking the F-box domain of Slmb to a nanobody recognizing GFP (VhhGFP4), significantly reduced fluorescent protein H2B-GFP in HeLa S3 cells overexpressing H2B-GFP with minimal off-target effects. This system can target and degrade functional GFP-tagged proteins to mimic protein loss, facilitating protein function and phenotype studies.
Baudisch et al. [141] extended deGradFP to plant research. They transformed tobacco plant cells with NSlmb-vhhGFP4 (targeted degradation peptide) and pGH219 (GFP expression) vectors. Western blot showed more pronounced GFP degradation by NSlmb-vhhGFP4 compared to NSnoFbox-vhhGFP4 control. This demonstrated fusion protein-based TPD for plant POI knockout via the ubiquitin-proteasome pathway, offering a novel protein modulation strategy in crop plants.
Ubox (CHIP)-Based Fusion Protein: Targeting c-Myc and KRAS
The RING E3 family has a unique subset, U-box, with a RING motif but lacking a Zn2+ binding site [39]. Like the RING domain, U-box engages E2 and facilitates substrate ubiquitination. CHIP is the most exemplary E3 ligase with a U-box domain.
c-Myc, a proto-oncogene product elevated in malignant tumors, forms a heterodimeric complex with the smaller basic helix-loop-helix/leucine zipper (bHLH/LZ) protein (Max), contributing to its cancer-promoting functions [142]. Max-U, the first U-box-based fusion protein, was constructed by Hatakeyama et al. [143], linking Max (a c-Myc binding motif) to the U-box region of CHIP (Fig. 2B). Max-U design verified c-Myc and Max interaction and enhanced c-Myc ubiquitination. Targeted c-Myc protein destruction by the artificial E3 was proven in vitro and in vivo.
Kirsten rat sarcoma viral oncogene homologue (KRAS) is a crucial therapeutic target for pancreatic, lung, and colorectal cancers. Raf-1, a key downstream KRAS effector, interacts with KRAS via two domains: Ras-binding domain (RBD) and Ras-associated domain (RAD) [144]. Based on this structure, Ma et al. developed a U-box-based fusion protein targeting KRAS for degradation [145]. The engineered E3 ubiquitin ligase, (RBD + CRD)Raf−1-U-Box (RC-U), harbors a KRAS recognition motif (RBD + CRD) conformally fused with the charged region and U-box domain of CHIP. Transfection of this fusion protein plasmid into PANC-1 cells with mutant KRAS significantly reduced KRAS levels.
VHL-EloB-EloC-CUL2-RBX1-Based Fusion Protein: AdPROM System
VHL is a key E3 ligase in PROTACs, with various peptide fragments and small molecules binding VHL [74, 76, 146]. However, E3 ligase, target protein junction discovery, and their interaction are often difficult and time-consuming. Rapidly identifying desired protein druggability properties in fusion protein TPD is advantageous. Fulcher et al. [147] established such a platform using an engineered VHL E3 ligase, termed AdPROM system (Fig. 2C). An anti-GFP nanobody (aGFP) was fused to either the N- or C-terminus of VHL to create VHL-aGFP. Transfecting cells with retroviruses encoding VHL-aGFP degraded endogenous GFP-tagged proteins via proteasomes. This platform facilitates protein function study and intracellular component sub-localization understanding. A limitation is that the affinity ligand itself may be recognized as an E3 substrate. Researchers proposed modifying AdPROM by replacing aGFP with smaller binders to specific endogenous proteins. They chose synthetic polypeptides called monobodies [148] recognizing Src-homology 2 domain-containing phosphatase 2 (SHP2), whose mutations are linked to aberrant Ras/MAPK activation and pathologies including cancers and Noonan syndrome. Two monobodies, aNSa1 (KD = 14 nM) and aCS3 (KD = 4 nM), selectively binding N-SH2 and C-SH2 domains of SHP2, were ligated to VHL to form VHL-aNSa1 and VHL-aCS3, respectively. Retroviral expression of VHL-aNSa1 or VHL-aCS3 reduced endogenous SHP2 in cells. The Ras/MAPK signaling pathway was also inhibited, shown by decreased ERK1/2 phosphorylation. This suggests synthetic monobodies can be used for AdPROM-mediated TPD.
STUbL RNF4-Based Fusion Protein: ARMeD System
RNF4, a relatively specific E3 ligase, has a C-terminal RING domain for dimerization and E2 recruitment, and an N-terminal domain with four small ubiquitin-like modifiers (SUMO) interaction patterns (SIM), allowing E3 ligase engagement of SUMOylated substrates. RNF4 is also known as a SUMO-targeted ubiquitin ligase, involved in cell growth and DNA damage response regulation of SUMOylated proteins like breast cancer type 1 susceptibility protein (BRCA1) and DNA damage checkpoint protein-1 (MDC1) [149, 150].
Ibrahim et al. [151] constructed an antibody RING-mediated destruction (ARMeD) system, replacing RNF4’s SUMO recognition domain with a substrate-specific nanobody. Anti-GFP nanobody (GNB) was used as a model, tethered to one or two RNF4 RING domains, creating GNB-1×RING and GNB-2×RING. Yellow fluorescent protein (YFP) fusion proteins (YFP-PARG and YFP-PML), theoretically recognized by GNB, were efficiently depleted in cells expressing doxycycline (Dox)-inducible GNB-RING constructs. Nanobodies (NNb2 and NNb9) targeting endogenous NEDD8 specific protease (NEDP1) were tethered to RNF4 RING to form Dox-inducible NNb-RING fusions (Fig. 2D). Apparent NEDP1 degradation and NEDD8 and dimer accumulation were seen. Proteomic analysis showed no observable off-target destruction, indicating ARMeD system high selectivity via high-affinity nanobodies. To bypass genetic manipulation and Dox induction, recombinant nanobody-RING fusion was introduced into cells via electroporation, leading to endogenous target protein elimination within minutes. This transient and rapid degradation method can study rapid cellular processes like the cell cycle.
SPOP-CUL3-RBX1-Based Fusion Protein: Optimized Degradation
Motivated by deGradFP by Caussinus et al. [140], Shin et al. [152] hypothesized that E3 architecture optimization could improve degradation. They constructed synthetic E3 ligase candidates fusing distinct E3 adaptor proteins to an anti-GFP nanobody (vhhGFP4). vhhGFP4-SPOP (Ab-SPOP), joining vhhGFP4 to SPOP (a CUL3-RING E3 ligase adaptor protein), optimally cleared H2B-GFP in cells, even compared to deGradFP.
Similarly, Lim et al. [153] systematically studied synthetic E3 ligases targeting GFP, termed bioPROTACs (biological PROTACs), validating their degradation effects in HEK 293 Tet-On 3G cells. Seven GFP binders (nanobodies: vhhGFP4, DARPin: 3G86, αReps: bGFP-A, bGFP-C, and three monobodies: GS2, GL6, GL8), and ten E3 ligases (βTRCP, FBW7, SKP2, VHL, SPOP, CRBN, DDB2, SOCS2, ASB1, CHIP) were used to explore bioPROTAC construction flexibility. Except for two weak binders, 5 of 7 GFP binders degraded GFP despite structural, size, and affinity diversity. Most vhhGFP4-E3 fusions degraded GFP, with SPOP showing greatest efficacy. 8 of 10 mammalian E3 ligases showed remarkable degradation activity. They also tested endogenous proliferating cell nuclear antigen (PCNA, a DNA polymerase δ auxiliary protein [154]) degradability via the proteasomal pathway using bioPROTAC. Rationally designed SPOP-con1, fusing the SPOP BTB domain to con-1 (a PCNA binding motif), efficiently induced PCNA degradation (Fig. 2E).
In 2021, Lim et al. [155] generated bioPROTACs against KRAS-GFP. SPOP was found to be the most suitable E3 ligase and was validated for RAS degradation by linking it to four high-affinity RAS binders (NS1, K27, K55, R11.1.6). SPOP-K27 showed complete pan-RAS degradation efficiency, degraded mutant KRASG12D, and inhibited KRAS-mutant AsPC-1 cell proliferation compared to other bio-degraders.
BioPROTACs are powerful for interrogating target biology, druggability, and TPD degrader creation. However, fusion protein-based bio-degraders are polar, lacking membrane permeability and bioavailability, requiring external cell entry methods (e.g., transfection, electroporation, membrane permeable peptides). Fusion proteins can complementarily screen suitable E3s, examine POI druggability, and lay the foundation for PROTAC advancement. With advanced gene delivery systems, fusion protein-based bioTPD is promising for clinical application.
Antibody (Fragments)-Based bioTPD: Harnessing Antibody Specificity
TRIM-Away: Antibody-Mediated Rapid Degradation
TRIM21, an E3 ligase, natively recognizes the Fc fragment of antibodies and drives antibody-POI complexes or antibody-bound pathogens to the proteasome [156, 157]. Trim-Away, an antibody-based bioTPD technology developed in 2017, exploits commercially available antibodies and TRIM21 for rapid protein disruption (Fig. 3A) [16]. Endogenous TRIM21 levels are sufficient for protein degradation in some cell types, like primary cells. If insufficient, TRIM21 needs exogenous delivery with antibodies via co-electroporation or microinjection. Trim-Away’s proof of concept was verified using 9 endogenous proteins in 10 cell types, showing broad application and substrate independence. Degradation is acute and rapid, within minutes. Using antibodies allows Trim-Away to target a wide range of POIs with commercially available antibodies and TRIM21 for functional studies. However, antibodies struggle to cross cell nuclei and membranes without external help. Clift et al. [16] showed Fc-nanobody fusion works with Trim-Away for nucleus protein degradation. In 2021, Chen et al. [158] created TRIMbody, fusing a POI-binding nanobody with the RBCC motif of TRIM21 to avoid microinjection or electroporation. Inducible EGFP TRIMbody expression efficiently degraded EGFP in HEK293T-EGFP cells via both proteasome and autophagy-lysosome pathways. Recently, two papers [159, 160] independently reported a BCL11A biological degrader proof-of-concept study based on Trim-Away. They produced nanobody plasmids fused to Trim21 or Fc, confirming selective BCL11A degradation via lentiviral transduction. Laura M. K. Dassamaf [160] designed a cell-permeant fusion of their nanobody to a cell-permeant miniature protein (ZF5.3) and an E3 adaptor (SPOP or RNF4). This fusion was expressed and efficiently depleted cellular BCL11A, offering a strategy for cell-permeant protein-based degraders.
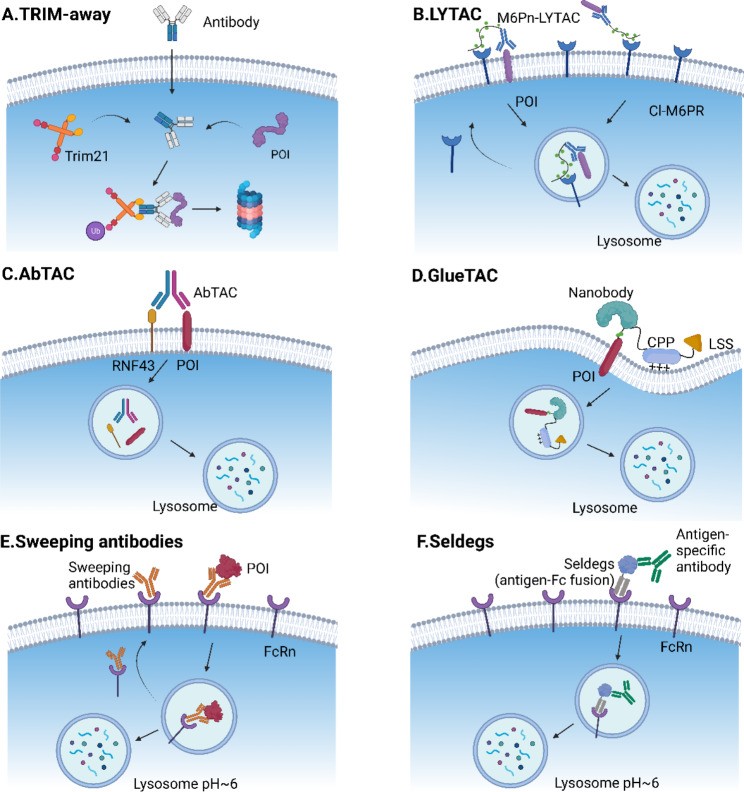
Fig. 3.
Open in a new tab
Fig. 3. Schematics illustrating various antibody-based bioTPD strategies, including Trim-Away, LYTAC, AbTAC, GlueTAC, Sweeping antibodies, and Seldegs.
Schematic representation of antibody-based bioTPD. (A) Trim21 recognizes the Fc domain of antibodies and is auto-ubiquitinated. Ubiquitinated Trim21 and its antibody/protein complexes are targeted for proteasomal degradation. (B) LYTAC is composed of a small molecule or an antibody coupled to a ligand that binds to LTRs, such as CI-M6PR and ASGPR. The LYTAC-POI complex is endocytosed along with LTR, followed by lysosomal degradation. (C) AbTAC, a bispecific antibody, concurrently recruits E3 ubiquitin ligases RNF43 and a membrane POI. The POI is degraded in a lysosomal-dependent manner. (D) GlueTAC consists of a covalent nanobody for POI targeting, a CPP for rapid endocytosis, and a lysosome-sorting sequence (LSS) for lysosomal degradation. (E) A sweeping antibody is an IgG that is engineered to connect to the neonatal Fc receptor (FcRn) at both neutral and acidic pH and a secreted POI only at neutral pH. The FcRn transports the POI-antibody-FcRn complex to the endosome. In the acidic environment, the POI leaves the sweeping antibody and proceeds to the lysosome, while the remained antibody-FcRn recycles back to the cell membrane to catch more targets (F) Seldegs are engineered Fc-antigen fusions with the capability to capture circulating antibodies and bring them to lysosomal degradation. The figure was created in BioRender.com
TRIM-Away has been practically applied in various cell types and in vivo embryo development [128, 161–165]. Unlike CRISPR/Cas9 and RNA interference, Trim-Away directly degrades specific proteins in any cell type, differentiating between splice or mutant protein variants and post-translationally modified proteins, opening new avenues for disease research.
LYTAC: Targeting Membrane and Extracellular Proteins to Lysosomes
Membrane-associated and extracellular proteins, encoded by 40% of genes [166], are vital in cancers, autoimmune disorders, and neurodegenerative diseases [167]. LYTAC complements PROTACs by selectively targeting these proteins to lysosomal degradation.
Lysosome-targeting receptors (LTRs) on the cell surface facilitate intracellular protein transport to lysosomes [168]. This process was used to create the first LYTAC, consisting of an antibody linked to an LTR-binding ligand. The first LYTAC used the cation-independent mannose-6-phosphate receptor (CI-MPR, IGF2R) as a lysosome shuttle [17]. The antibody targets POI, and conjugated multiple serine-O-mannose-6-phosphonate (M6Pn) residues interact with CI-MPR for clathrin-mediated endocytosis, dragging POI and LYTAC to lysosomes for degradation (Fig. 3B). This LYTAC platform degrades plasma proteins (apolipoprotein E4) and membrane proteins, including EGFR, transferrin receptor-1 (TfR/CD71), and PD-L1.
LYTACs targeting tissue-specific LTRs can induce POI degradation in specific tissues. Unlike ubiquitous CI-MPR, some LTRs are tissue-specific. Asialoglycoprotein receptor (ASGPR), primarily expressed in hepatocytes (~500,000 copies per cell), is a well-defined LTR [169]. ASGPR-based LYTACs (GalNAc-LYTACs) are created by fusing antibodies or peptides with N-acetyl galactosamine (GalNAc) or tri-GalNAc as ASGPR ligands [18, 170, 171]. In liver cancer cells, GalNAc-LYTACs downregulate EGFR and integrins and internalize extracellular components better than M6Pn-LYTACs in HEPG2 cells, likely due to higher ASGPR than CI-MPR levels in hepatocytes [18]. CI-MPR- and ASGPR-based LYTAC success suggests exploiting other cell-specific and tissue-specific LTRs [10]. Current LYTACs are degraded with POIs, lacking catalytic function unlike most PROTAC degraders [45]. Antibody size and immunogenicity of conjugated glycopeptides need to be addressed.
AbTAC: Bispecific Antibodies for Membrane Protein Degradation
Bispecific antibodies, recombinant antibodies recognizing two antigens or epitopes, are rapidly growing in cancer immunotherapy [172]. In 2021, Wells’ group [19] used AbTAC, a bispecific antibody, to recruit E3 ubiquitin ligases RNF43 and PD-L1. RNF43 is a transmembrane E3 ligase with an intracellular RING domain and a structured ectodomain [173]. AbTAC was created by fusing two half IgGs targeting PD-L1 and the RNF43 ectodomain (Fig. 3C). Biolayer Interferometry (BLI) confirmed high affinity for both antigens. AbTAC efficiently depleted PD-L1, with a half-maximal degradation concentration (DC50) of 3.4 nM and 63% maximum degradation efficacy at 24 hours in MDA-MB-231 cells. Unexpectedly, AbTAC depleted PD-L1 in a lysosome-dependent, not proteasome-dependent, manner, similar to LYTAC. AbTAC’s exact mechanism needs further study. Like LYTACs, AbTAC treatment caused no large cellular proteomic perturbations. Recently, Wells’ group [174] created a new AbTAC system co-opting another transmembrane E3 ligase, zinc and ring finger 3 (ZNRF3), to disrupt EGFR and PD-L1, showing that antibody binding epitopes on E3 ligase and POI are more important than AbTAC affinities. A similar approach, bispecific proteolysis-targeting antibodies (PROTABs), tethers cell-surface E3 ubiquitin ligases (RNF43, ZNRF3) to transmembrane proteins (insulin growth factor 1 receptor (IGF1R)) [175]. PROTAB induces ligase-dependent IGF1R internalization and degradation, demonstrating this platform’s generality for human epidermal growth factor receptor 2 (HER2) and PD-L1 degradation. Given that RNF43 and ZNRF3 are downstream of Wnt signaling, PROTABs can target Wnt-hyperactivated tumors and specifically degrade cell-surface proteins.
Despite proteasome-based TPD advances, only cytosolic E3 ligases have been used. These studies first extended proteasomal degradation to cell-surface E3 ligases, offering more membrane-bound protein targeted degradation methods.
GlueTAC: Covalent Nanobody-Based Lysosomal Targeting
Unlike other antibody-based TPDs (e.g., LYTAC, AbTAC), Zhang et al. [20] developed GlueTAC, targeting cell-surface proteins via covalent nanobody-PROTAC. They screened a PD-L1-targeted covalent nanobody variant (Gluebody) using MS-assisted screening platform (MSSP) with genetic code expansion (GCE). Covalent nanobodies improve cell penetration, binding affinity, and reduce off-target effects via covalent interaction with POI. GlueTAC was coupled to CPP (GGGRRRRRRRRR) and lysosome-sorting sequence (NPGY) for rapid endocytosis and lysosomal degradation [176, 177] (Fig. 3D). GlueTAC efficiently degraded cellular PD-L1, showing superior antitumor activity in the PD-L1-EGFP/A375 tumor model compared to Atezolizumab, an FDA-approved anti-PD-L1 antibody.
Compared to LYTAC or AbTAC, GlueTAC is a universal membrane protein targeted degradation strategy, cell-type and receptor/E3 ligase-independent. However, safety concerns from unnatural amino acids and nanobody pharmacokinetics need consideration.
Sweeping Antibodies: Recyclable Degraders for Extracellular Antigens
Sweeping antibodies are recyclable degraders specifically targeting extracellular antigen degradation, reducing traditional antibody dose and administration frequency [178]. Sweeping antibodies are pH-dependent bispecific IgGs engineered to bind neonatal Fc receptor (FcRn, a recycling receptor) at neutral/acidic pH and secreted/soluble proteins only at neutral pH. FcRn, a membrane receptor for IgG and albumin, prolongs lifespan and maintains dynamic balance of these proteins [179, 180]. FcRn transports POI-antibody-FcRn complex to the endosome. In the acidic environment, POI leaves the sweeping antibody and proceeds to the lysosome, while the antibody-FcRn complex recycles back to the cell membrane (Fig. 3E).
Igawa et al. [22] first constructed a sweeping antibody by manipulating the variable region for pH-dependent binding and modifying the constant region to improve FcRn affinity for internalization. An anti-interleukin-6 receptor (IL-6R) sweeping antibody derived from tocilizumab cleared plasma IL-6R 50- to 1000-fold in mice compared to a conventional antibody. Muramatsu et al. [181] designed myostatin-specific sweeping antibodies to reinforce muscle strength by sweeping latent myostatin. Sampei et al. [182] engineered a pH-dependent antibody specific to complement component 5 (C5), demonstrating long-lasting C5 clearing activity in cynomolgus monkeys, suggesting application in modulating disordered complement systems.
Seldegs: Selective Degradation of Autoantibodies
Seldegs, engineered antibody fragment-antigen fusion proteins, selectively deplete endogenous antigen-specific immunoglobulin G (IgG) based on FcRn-IgG interactions. These clearing agents offer promise for antibody-mediated autoimmunity disorders, transplant rejection, and IgG-drug complex clearance [183].
Like sweeping antibodies, Seldegs mainly comprise an engineered Fc domain targeting FcRn. Seldegs evolved from earlier Abdegs. In 2015, Ward’s group [183] engineered the Fc fragment of human IgG to increase affinity and mitigate pH dependence on FcRn, naming it Abdegs (antibodies that enhance IgG degradation), inducing rapid in vivo clearance of unmanipulated circulating IgG. As Abdegs non-specifically degraded all circulating IgGs, they generated selective clearing agents in 2017, Seldegs (selective degradation) [23]. Seldegs are Fc-antigen fusion proteins capturing circulating antibodies and targeting them for lysosomal degradation based on high pH-independent Seldeg-FcRn interactions (Fig. 3F). Specific mutations were introduced into the Fc domain to ablate FcγR affinity and increase FcRn affinity [184]. Myelin oligodendrocyte glycoprotein (MOG) and HER2 antigens were fused with the mutated Fc domain. MOG- and HER2-Seldegs induced lysosomal delivery of anti-MOG and anti-HER2 antibodies in FcRn-expressing cells and in vivo clearance of targeted antibodies at low doses, unlike Abdegs or earlier FcRn antagonists with no effect on total IgG levels [183, 185, 186]. Further investigation verified that MOG-Seldeg treatment specifically removed patient-derived MOG antibodies, ameliorating autoimmune encephalomyelitis symptoms in mice [187]. As Seldegs contain recombinant antigen, it must be ensured that the antigen only binds targeted autoantibodies, not affecting non-target antibodies [188].
Nucleic Acid-Based bioTPD: Expanding Target Range to RNA and Transcription Factors
Proteins with catalytic activity are often druggable. However, many protein families like RNA binding proteins (RBPs) and transcription factors (TFs) remain intractable due to lacking binding sites. Specific binding ligand discovery is critical to develop drugs targeting such proteins. Nucleic acids can bind specific protein domains, making them powerful biomacromolecule ligands for degrader creation. Nucleic acid-based degraders have rapidly evolved since 2021, with TF PROTACs, RNA-PROTACs, G-quadruplex (G4)-PROTACs, and aptamer-based PROTACs developed within a year, offering potential to directly target diseases caused by RBPs, TFs, or G4-binding proteins.
RNA-PROTACs: Degrading RNA Binding Proteins
RBPs are a large class of >2,000 proteins interacting with transcripts in most RNA-driven processes [189]. RBPs bind RNAs dynamically and sequence-dependently, forming ribonucleoprotein (RNP) complexes and coordinating RNA processing [190, 191] (Fig. 4A). RBP genetic alterations can cause genetic diseases, including amyotrophic lateral sclerosis from fused-in-sarcoma (FUS)/TAR DNA binding protein-43 (TDP-43) mutations, and myelodysplastic syndromes from U2AF35/ splicing factor 3b subunit 1 (SF3B1) mutations [192]. However, most RBPs are undruggable by conventional therapies or small-molecule PROTACs [193].
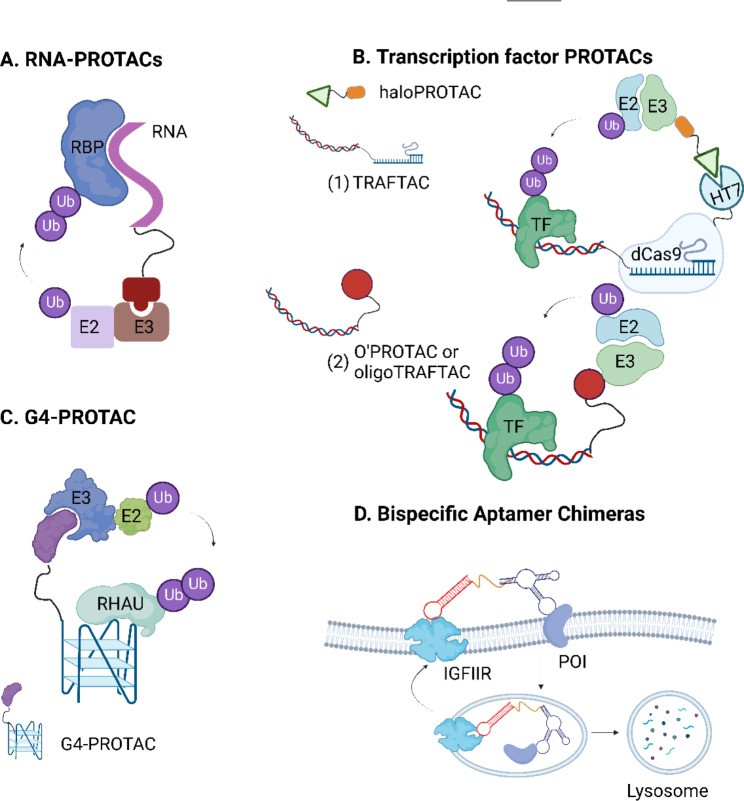
Fig. 4.
Open in a new tab
Fig. 4. Illustration of nucleic acid-based bioTPD strategies, including RNA-PROTACs, TRAFTACs, G4-PROTACs, and Bispecific aptamer chimeras.
Schematic representation of nucleic acid-based bioTPD. (A) RNA-PROTAC consists of a short oligonucleotide that is iso-sequential with the RNA consensus binding element as an RBP-recognizing ligand and an E3-recruiting peptide for proteasomal degradation. (B) TRAFTAC (1) is a bifunctional chimeric oligonucleotide (dsDNA-CRISPR-RNA) that binds to the transcription factor with an oligonucleotide and recruits E3 ligases through dCas9-HT7 fusion protein in the presence of a haloPROTAC. O’PROTAC or OligoTRAFTAC (2) contains a double-stranded oligonucleotide as a transcription factor-recognizing ligand and a VHL-recruiting moiety. (C) G4-PROTAC uses G4 as a warhead of the PROTAC for targeted degradation of a G4-binding protein RHAU (a DEAH-box helicase). (D) Bispecific aptamer chimeras utilize DNA aptamers to target the POI and lysosome-shuttling receptor IGFIIR, respectively. The figure was created in BioRender.com
In 2021, Ghidini et al. [194] introduced RNA-PROTAC, chimeric structures degrading RBPs. RNA-PROTACs use short oligonucleotides iso-sequential with the RBP RNA consensus binding element as RBP-recognizing ligands, linked to an E3-recruiting peptide. Proof-of-concept was confirmed by targeting stem cell factor LIN28 and splicing factor RBFOX1, with rationally designed chimeras selectively degrading these RBPs in cancer cell lines in a ubiquitin-dependent manner.
Transcription Factor PROTACs: Targeting Gene Expression Regulators
TFs are DNA-binding proteins regulating gene expression, with dysfunction causing pathologies including cancers [195]. TFs lack active or allosteric sites found in kinases or enzymes, making TF-binding small-molecule inhibitors difficult to design, thus TFs are poorly druggable targets.
Using TF DNA-binding ability, Crews’ group [196] developed TRAnscription Factor Targeting Chimeras (TRAFTACs) to induce TF degradation. TRAFTACs are bifunctional chimeric oligos (dsDNA-CRISPR-RNA) bridging the TF of interest (TOI) with ectopically expressed dCas9-Halotag7 fusion protein (dCas9HT7) to form a complex. HaloPROTAC incubation recruits VHL-E3 ligase to the complexed fusion protein, inducing TOI deconstruction (Fig. 4B). TRAFTACs successfully degraded oncogenic TOIs NF-κB and brachyury via the proteasomal pathway. They also degraded zebrafish brachyury, inducing a no-tail phenotype, suggesting therapeutic potential for degrading disease-relevant TFs. Later, Crews’ group [194] developed second-generation TRAFTACs, oligoTRAFTAC, succinctly consisting of a TF binding oligonucleotide and an E3 ligase-recruiting ligand without genetic modification (Fig. 4B). Two specifically designed oligoTRAFTACs effectively degraded c-Myc and brachyury in cells and zebrafish.
Shao et al. [197] reported similar oligonucleotide PROTACs, O’PROTACs, also containing double-stranded oligonucleotides as TF-recognizing ligands and a VHL-recruiting moiety (Fig. 4B). O’PROTACs degraded oncogenic TFs lymphoid enhancer-binding factor 1 (LEF1) and ETS-related gene (ERG), showing suppressive effects in prostate cancer models. OligoTRAFTAC and O’PROTAC exclude artificially engineered dCas9-HT7 fusion protein, simplifying synthesis and improving nucleic acid-based bioTPD clinical application limitations compared to first-generation TRAFTACs.
Unlike oligoTRAFTACs and O’PROTACs, Liu et al. [198] used click reactions to connect DNA oligonucleotides to E3 ligase ligands. They synthesized and optimized VHL-based TF-PROTAC series (dNF-κB and dE2F) by changing linker length and structure via copper-free strain-promoted azide-alkyne cycloaddition (SPAAC) reaction. These TF-PROTACs selectively degraded p65 and E2F1 protein in cells, showing promising antiproliferative effects.
DNA-based PROTACs are more stable than RNA-PROTACs. DNA binding specificity to TFs is also better than RNA binding specificity to RBPs [198].
G4-PROTAC: Targeting G-Quadruplex Binding Proteins
G4 binding proteins are involved in important biological processes like telomere maintenance, DNA replication, and gene transcription [199]. G4 binding protein abnormalities are linked to human diseases like cancers and amyotrophic lateral sclerosis (ALS) [200]. G4s are four-stranded DNA secondary structures with rich guanine sequences [200].
Patil et al. [201] first used G4 as a PROTAC warhead for targeted degradation of G4-binding protein RHAU (DEAH-box helicase) (Fig. 4C). RHAU is overexpressed in C9orf72-related ALS patients, making it a vital ALS treatment target [202]. An RHAU-bound all-parallel-stranded G4 (sequence TT(GGGT)4) was linked to CRBN and VHL E3 ligands via click reaction. Both G4-PROTACs showed potent and specific RHAU degradation in HeLa and K-562 cell lines, highlighting the feasibility of TPD construct design using non-canonical nucleic acid structures and offering alternative therapeutics for diseases caused by G4-binding proteins.
Bispecific Aptamer Chimeras: Lysosomal Targeting of Membrane Proteins
Aptamers are short single-stranded oligonucleotides (ssDNA or ssRNA) selectively binding protein targets or peptides with high affinity, either in native states or on cellular membranes [203]. Aptamers can be screened in vitro via systematic evolution of ligands by exponential enrichment (SELEX), broadening aptamer target range to all accessible proteins [204–206]. Nucleic acid ligands can effectively target proteins for proteasomal and lysosomal degradation.
Inspired by LYTAC design, which uses cell-surface lysosome-shuttling receptors to direct membrane proteins to lysosomes [17], Miao et al. [21] designed bispecific aptamer conjugates targeting Insulin-like growth factor type II receptor (IGFIIR, a lysosome-shuttling receptor) and membrane POIs (Fig. 4D). These chimeras shuttled mesenchymal epithelial transition (Met) and tyrosine protein kinase-like 7 (PTK-7) to lysosomes, rapidly and efficiently degrading them at nanomolar concentrations. SELEX/Cell-SELEX for aptamer selection [206, 207] might allow more membrane protein-associated degradation than LYTAC. However, aptamer chimera stability and off-target effects need further study [208].
Options for Improving the Delivery Efficacy of bioTPD: Overcoming Delivery Hurdles
Table 1 shows poor cell permeability, unfavorable pharmacokinetics, low stability, and low delivery efficacy are common bio-degrader limitations. While bioTPD has potential as highly specific therapeutic agents, delivery and degradation efficiency are key hurdles.
Utilization of CPPs: Enhancing Cell Entry
Cell-penetrating peptides (CPPs), including oligoarginine (RRRRRRRR), HIV-1 Tat peptide (YGRKKRRQRRR), pentapeptide (RRRRK), and Xentry (LCLRPVG), facilitate cell entry via direct penetration or endocytosis, delivering peptides, oligonucleotides, proteins, and nanocarriers [104, 110, 209–211]. Several studies use CPPs to improve bioTPD cell permeability. For example, poly-D-arginine motifs were incorporated into phosphoPROTACs coupling tyrosine phosphorylation sequences with VHL-recruiting peptides for cell permeability [110]. TAT-modified hydrophobic tags conjugating peptides for TAR DNA binding protein 43 (TDP-43) disruption reduced TDP-43-induced cytotoxicity in N2a cells [210]. RRRRK sequence also enhanced cell permeability of CREPT-targeted PROTAC in AsPc-1 and MIA PaCa-2 cells, comparable to TAT action [104]. CPPs also facilitate cell entry of antibody-based cargo, as seen in GlueTAC. To improve GlueTAC cell entry and proteolytic capabilities, a CPP peptide of nine D-arginines was tethered between the C-terminal of an anti-PD-L1 nanobody and the N-terminal of a lysosomal sorting sequence [20].
Improve Selectivity by Active Targeting Ligands: Precision Delivery
Selectivity to the POI is crucial for therapeutics. High selectivity reduces off-target effects and prevents side effects. Improving selectivity is a constant challenge for TPD.
E3 ligase selection is important for improving selectivity, as E3 ligase expression varies in tissues, cells, and subcellular compartments [212]. Linker length optimization and ternary complex stabilization can also enhance PROTAC selectivity beyond parent ligands [213]. However, computationally predicting optimal linkers and lengths without POI-PROTAC-E3 ternary complex structures is challenging [212].
Active targeting ligands, like antibodies or aptamers, can enhance TPD selectivity, potentially targeting diverse tissues or cell types. Antibody-PROTAC conjugates (Ab-PROTACs), analogous to antibody-drug conjugates (ADCs), have emerged. Anti-HER2 antibodies were joined to BRD4 or estrogen receptor alpha (ERα) degraders [214, 215]. These Ab-PROTACs showed specific internalization and strong POI degradation in HER2-positive cells. Dragovich et al. [216] systematically constructed antibody-PROTAC conjugates tethering a BRD4 degrader to anti-STEAP1 or anti-CLL1 antibodies, showing that the linker between antibody and PROTAC and its cleavable property significantly impacted degradation efficacy.
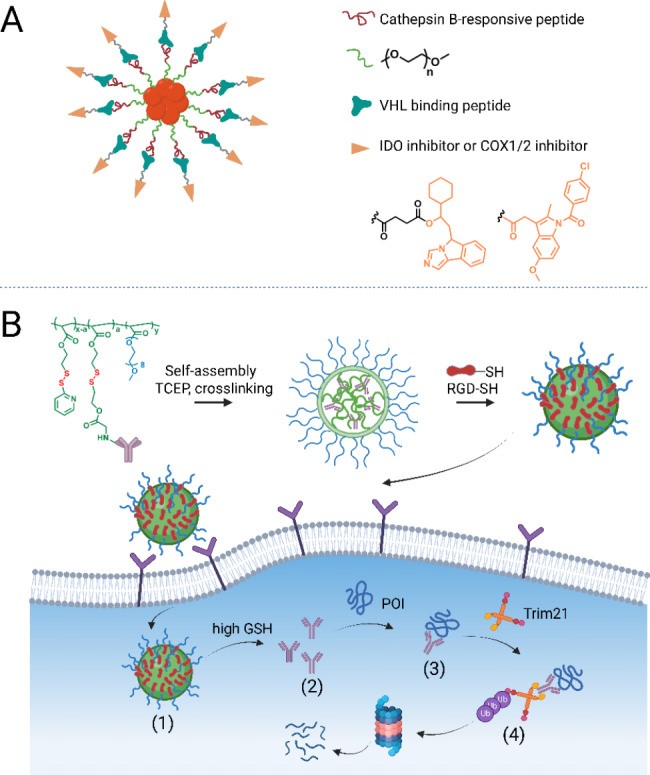
Aptamer-PROTAC conjugates, recently developed, conjugate a bromodomain and extra-terminal (BET) degrader to nucleic acid aptamer AS1411 via a GSH-responsive linker [218]. Aptamer AS1411, a transport agent for cell-surface nucleolin-expressing cancer cells, improved degrader uptake and internalization in nucleolin-overexpressing MCF-7 cells, leading to high in vivo BRD4 degradation, antitumor potency, and decreased toxicity. Aptamer conjugation may be advantageous for PROTAC delivery, similar to antibody conjugation. These studies demonstrate proof-of-concept for tissue/cell-specific target degradation, overcoming PROTAC selectivity constraints, with significant potential for other TPD types, including bioTPD.
Drug Delivery Systems: Nanocarriers for Enhanced BioTPD
Drug delivery systems have rapidly advanced, offering unique advantages in delivering therapeutic agents with undesirable properties. Drug delivery nanocarriers, with broad loading capacity, improved pharmacokinetics/pharmacodynamics, accelerated cellular uptake, and multifunctional modification, may play a vital role in bioTPD clinical translation by maximizing efficacy and overcoming limitations [219].
Researchers are using drug delivery systems to deliver small-molecule chimeric degraders and bioTPD components to improve in vivo applications. Small-molecule PROTAC nanoformulations include polymeric nanoparticles [219, 220], lipid-based nanoparticles [221], biomimetic nanocarriers [222], and two-dimensional nanocarriers [223] to improve destruction efficacy. For example, PLGA-PEG nanoparticles encapsulated hydrophobic ARV-825, achieving passive targeting and enhanced in vivo antitumor effect [220]. ARV-825-loading substance P (SP) peptide-modified polymeric micelles promoted ARV-825 movement across the blood-brain barrier, showing outstanding anti-proliferative effects against glioma [219], opening glioma therapy avenues using TPD. Delivery systems are suitable for small-molecule TPD delivery. BioTPD delivery demands vary based on bioTPD types.
Peptide-based degraders have poor cell permeability, low stability, and unfavorable pharmacokinetics. Nano-delivery can overcome these bottlenecks. Zhang et al. [224] reported nano-PROTACs conjugating a small-molecule-peptide PROTAC to a semiconducting polymeric nanoparticle via a cathepsin B-susceptible fragment (Fig. 5A). The indoleamine 2,3-dioxygenase (IDO)-targeting PROTAC comprised a VHL-binding peptide and IDO inhibitor (NLG919). Nano-PROTACs integrated phototherapeutic effects and controllable protein degradation for photo-immunometabolic anticancer therapy. Polymeric construct design prolonged circulation time and accumulated in tumors via enhanced permeability and retention (EPR) effect. Nanoparticles showed excellent cellular uptake. After tumor cell entry, PROTAC molecules were released by cathepsin B, inducing IDO elimination via the proteasomal pathway, boosting antitumor T-cell immunity in 4T1-bearing mice. They used a similar polymeric nanosystem to deliver cyclooxygenase 1/2 (COX1/2)-targeting PROTAC, reshaping the immunosuppressive tumor microenvironment and reinforcing anticancer immunotherapy [225], corroborating drug delivery systems can improve peptide-based bioTPD drug-like properties.
Fig. 5.
Open in a new tab
Fig. 5. Visual representation of drug delivery systems enhancing bioTPD efficacy, including polymeric nanoparticles and antibody-conjugated nanogels.
Drug delivery system to improve the delivery efficacy of bioTPD. (A) A polymeric nanoparticle-mediated delivery system was used for combination therapy of phototherapy, and PROTAC-engaged immunotherapy. (B) The antibody-tethered Nano-ERASER was transported into cells (1) and subsequently released antibodies upon a high level of glutathione (GSH) (2). The antibodies primed the corresponding POI (3) and drive them for degradation through Trim-Away pathway (4). The figure was created in BioRender.com
TRIM-Away is a rapid, highly selective bioTPD platform [16], but antibody cell penetration is poor. Microinjection or electroporation, used to overcome this, are unfavorable for clinical translation. In 2021, Sui et al. [226] developed Nano-ERASER, a practical, secure Trim-Away version, engrafting antibody-conjugated polymer nanogel to deliver and release antibodies in the cytosol, as an alternative to microinjection and electroporation. Antibody-tethered Nano-ERASER was transported into cells via receptor-mediated endocytosis, releasing antibodies upon high glutathione (GSH) levels (Fig. 5B). GFP and coatomer protein complex ζ1 (COPZ1) were successfully eliminated via Trim-Away, paving the way for in vivo and clinical translation of membrane-impermeable antibody-based bioTPD.
In Vitro Selection of Binders for bioTPD: High-Throughput Screening Technologies
bioTPD design begins with identifying a high-affinity binder for the target. Rapid, high-throughput screening techniques for extensive high-specific binder repertoires are important for bioTPD development. Common binders are peptides, antibodies, and antibody fragments. In vitro display technologies are powerful for selecting peptides and antibodies from constructed libraries. Table 3 outlines frequently used selection platforms for peptide/antibody screening.
Table 3. Overview of display technologies
| Screening technologies | Display principle | Selection principle | Typical library size |
|---|---|---|---|
| Phage display | Protein/peptide library is displayed on the surface of phage particles | Capture/elution | 109–1011 |
| Cell-surface display | Protein/peptide library is displayed on the surface of a living cell as a fusion to cell surface protein | FACS selection | 107 eukaryotes 108–1010 prokaryote |
| Ribosome display | Protein/peptide library is displayed on the stalled ribosome/mRNA complex | Capture/elution | 1012–1014 |
| mRNA/cDNA display | Protein/peptide library is displayed as covalently attached peptides to its encoding mRNA/cDNA | Capture/elution | 1012–1014 |
Open in a new tab
Table 3. A comparison of different display technologies used for binder selection, highlighting their principles and library sizes.
Phage display, the first in vitro display technology, presents exogenous proteins/peptides on bacteriophage surfaces as fused proteins with phage coat proteins [227]. Phage display principle is physical linkage between phage phenotype and genotype, allowing foreign protein information retrieval based on inserted genes [228]. Phage display screening involves library construction (huge DNA clones carrying genes encoding peptides or antibody fragments), cloning these genes into the phage genome, and producing phage particles displaying foreign proteins in E. coli. The phage library is then selected via biopanning, incubated with immobilized antigen, and bound phage clones with target molecules are collected, amplified, and subjected to 2–4 biopanning rounds to enrich high-affinity phage clones [229]. High-affinity phage clones are selected, and inserted genes encoding foreign proteins are sequenced and analyzed to obtain peptide or antibody fragment sequence information. Phage display is a versatile tool to identify specific antigen binders due to its simplicity and efficacy.
Cell-surface display and cell-free display technologies are also used [229, 230]. Cell-surface display technologies have been developed in yeast, bacteria, and mammalian cells, expressing proteins or peptides on their surface for ligand screening, with hosts replicating autonomously, offering advantages over phages (complex mammalian protein production, post-translational modifications) [231, 232]. However, cell-based display limitations are smaller library sizes and specialized sorting equipment [233].
Cell-free display technologies include ribosome (mRNA) and cDNA display [234], using similar screening processes to phage display (binding, washing, elution, amplification) [235]. Cell-free display allows library sizes exceeding 1013 variants, orders of magnitude larger than other display technologies [234, 236].
Outlook: The Future of bioTPD and Targeted Therapeutics
Given the progress and attention from academia and industry over the past two decades, TPD is a highly promising therapeutic modality. As Crews et al. stated, “The past is prologue” best describes the current TPD landscape. New TPD platforms like LYTAC, AbTAC, and Trim-away have generated significant excitement around biological TPD technologies, poised to revolutionize classical PROTACs by modulating more undruggable targets. Numerous conceptual TPD designs are continuously being developed, opening novel clinical application avenues. bioTPD development is still in early exploratory stages, requiring further studies to address limitations. Future efforts should focus on elucidating degradation mechanisms for new TPD concepts and accelerating bioTPD clinical translation. Much like specialized tools – perhaps something akin to das xentry hgb 99 – are essential for navigating the complexities of modern automotive diagnostics, continued innovation and refinement in bioTPD are crucial for unlocking its full therapeutic potential and addressing the intricate challenges of human disease.
Acknowledgements
Not applicable.
Abbreviations
α-syn Alpha-synuclein.
AbTAC Antibody-based PROTAC.
AdPROM Affinity-directed protein missile.
AID Auxin-inducible degron.
Akt Serine/threonine-protein kinase AKT.
APC Allophycocyanin.
AR Androgen receptor.
ARMeD Antibody RING-mediated destruction.
ASGPR Asialoglycoprotein receptor.
ATTEC Autophagy-tethering compounds.
AUTAC Autophagy-targeting chimera.
AUTOTAC AUTOphagy-TArgeting Chimera.
BioPROTAC Biological PROTAC.
BIR3 baculovirus IAP repeat 3.
BRD4 Bromodomain-containing 4.
CDK5 Cyclin-dependent kinase 5.
CHAMP Chaperone-mediated protein degradation.
CHIP Carboxyl-terminal Hsp70 interacting protein.
CI-M6PR Cation-independent mannose-6-phosphate receptor.
CMA Chaperone-mediated autophagy.
CMPD Cell membrane penetrating structural domain.
CPP Cell penetrating peptide.
CRBN Cereblon.
CREPT Cell cycle-related and expression-elevated protein in tumor.
CRISPR Clustered regularly interspaced short palindrome repeats.
CTM CMA targeting structural domain.
DAPK1 Death-associated protein kinase 1.
DARPin Designed ankyrin repeat proteins.
DC50 Half-maximal degradation concentrations.
ERG ETS-related gene.
ERα Estrogen receptor alpha.
ERβ Estrogen receptor beta.
FcRn Neonatal Fc receptor.
FKBP FK506- and rapamycin-binding protein.
GalNAc N-acetyl galactosamine.
HIF Hypoxia-inducible factor.
HyT Hydrophobic tag.
IAP Inhibitor of apoptosis protein.
IGFIIR Insulin-like growth factor type II receptor.
IgG Immunoglobulin G.
ITC Isothermal titration calorimetry.
KD Dissociation constant.
KEAP1 Kelch-1ike ECH- associated protein l.
KRAS Kirsten rat sarcoma viral oncogene homologue.
L2A Lysosome-associated membrane protein type 2 A.
LBs Lewy bodies.
LEF1 lymphoid enhancer-binding factor 1.
LID Ligand-Induced Degradation.
LSS Lysosomal sorting sequence.
LYTAC Lysosome-targeting chimeras.
MADTAC Macroautophagy Degradation Targeting Chimeras.
MDM2 Mouse double minute-2.
Met Mesenchymal epithelial transition.
MetAP2 Methionine aminopeptidase 2.
MIF Macrophage migration inhibitory factor.
MOG Myelin oligodendrocyte glycoprotein.
NF-κB Nuclear factor kappa-light-chain enhancer of activated B cells.
NR2B N-methyl-D-aspartate receptor 2B.
Nrf2 Transcription factor NF-E2-related factor 2.
PBD Target protein binding structural domain.
PCC Protein-catalyzed capture.
PCNA Proliferating cell nuclear antigen.
PD-1 Programmed cell death 1.
PDAC Pancreatic ductal adenocarcinoma.
PD-L1 Programmed cell death ligand 1.
PI3K Phosphatidylinositol-3-kinase.
PIPK2 Receptor Interacting Serine/Threonine Kinase 2.
PK Pharmacokinetics.
POI Protein of interest.
PROTAC PROteolysis-TArgeting Chimeras.
PTK-7 Protein kinase-like 7.
RIBOTAC Ribonuclease targeting chimera.
RING Really Interesting New Gene.
RNF4 Ring Finger Protein 4.
SCF SKP1, CUL1, and F-box protein.
SHP2 SH2-containing protein tyrosine phos-phatase 2.
SMPI Small molecule protein hydrolysis inducer.
SNIPERs Specific and Non-genetic IAP-dependent Protein Erasers.
SPOP Speckle-type BTB/POZ protein.
SUMO Small ubiquitin-like modifier.
Tau microtubule-associated protein.
TBK1 TANK-binding kinase 1.
TPD Targeted protein degradation.
TRAFTAC TRAnscription Factor Targeting Chimeras.
TRIM21 Tripartite motif-containing protein 21.
TRIM24 Tripartite motif containing 24.
VHL Von Hippel-Lindau.
Authors’ contributions
LY, ZJL, JHW and JGW directed the preparation of this review and designed the structure of this manuscript. HFW, RHZ, FSX and KJY wrote the manuscript. RHZ and LHZ produced the figures, and PZ and GWS prepared the tables in this review. LYD and CCX revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from GuangDong Basic and Applied Basic Research Foundation (2021A1515012164, 2022A1515010943), International Science and Technology Cooperation for Shenzhen Technology Innovation Plan (GJHZ20200731095411034), National Postdoctoral Science Foundation of China (2022M712189), National Key Research and Development Program of China (2020YFA0908000), Innovation Team and Talents Cultivation Program of the National Administration of Traditional Chinese Medicine (ZYYCXTD-C-202002), National Natural Science Foundation of China (82074098, 81841001), Shenzhen Science and Technology Innovation Commission (JCYJ20210324115800001 and JCYJ20210324114014039), National Natural Science Foundation of China (32000026, 32170774), Open Research Funds of the State Key Laboratory of Ophthalmology (2022KF06), Shenzhen Science and Technology Program (JCYJ20210324113608023), National Natural Science Foundation of China (32101219), and Shenzhen Science and Technology Innovation Committee (RCBS20210706092213007).
Data availability
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of Competing Interest
The other authors declare no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally to this work: Huifang Wang, Runhua Zhou and Fushan Xu.
Contributor Information
Le Yu, Email: [email protected].
Zhijie Li, Email: [email protected].
Jianhong Wang, Email: [email protected].
Jigang Wang, Email: [email protected].
References
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.